STRipy application
STRipy main graphical application
STRipy is an application for detecting short tandem repeats (STRs) from Illumina short-read sequencing data. Whole genome, whole exome and targeted aligned sequencing files are acceptable, as long as there is coverage on the targeted STR locus. STRipy's main application is divided into two: 1) the Client which is the main graphical application a user interacts with, and 2) the Server where genotyping takes place. We have set up a publically accessible server that is running on Cloud which can be used by everyone. Therefore, one does not need to set up a server (download and set up tools and reference genomes) and can easily use the Cloud for analysing samples.
STRipy pipeline version
While STRipy's main graphical application is good for genotyping few samples and loci at a time, then it is not suitable for a large-scale analysis. We have created a non-graphical version of STRipy (STRipy-pipeline) that can be run from the command line and which is easy to implement into clinical pipelines. This version also uses parallel computing for faster genotyping and in the end, a HTML report (and/or a JSON file) is created matching the genotypes with the information in our STRs database (see an example report here). The command-line/pipeline version also supports custom coordinates via BED file. This not only enables the genotyping any STR locus in the genome, but also VNTRs. We have only validated the known pathogenic STR loci and therefore the accuracy of genotyping custom ones is not known.
Download, source code and set-up instructions
STRipy's Client can be downloaded as a stand-alone application from the front page or you can run the app from the source code (gitlab.com/andreassh/stripy-client). In order to run your own server, download STRipy's Server and follow the directions at its repository (gitlab.com/andreassh/stripy-server). A non-graphical command-line version of STRipy is available from gitlab.com/andreassh/stripy-pipeline which can be integrated into clinical pipelines. STRipy can run under macOS, Linux and Windows 10/11 through WSL.
Please see the STRipy's documentation page for installation and set-up instructions.
Method
Short overview
STRipy uses ExpansionHunter (Dolzhenko, et al., 2017) to determine genotypes and REViewer (Dolzhenko, et al., 2021) for visualizing read alignments. From a sequencing file, STRipy extracts out a small fraction of reads that are relevant to the targeted locus and coordinates of mis-mapped reads (if they exist). The output data is then forwarded to specialised tools for genotyping and creating read visualisations.
When using the main application, reads that were extracted out are sent to the Server (either Cloud or a local computer/internal network). For privacy purposes, the file name of your sample will be anonymised before sending it to the Cloud and all files analysed on Cloud will be deleted immediately after genotyping. Information about users (such as IP addresses) or samples (extracted reads, results, etc.) will not be stored.
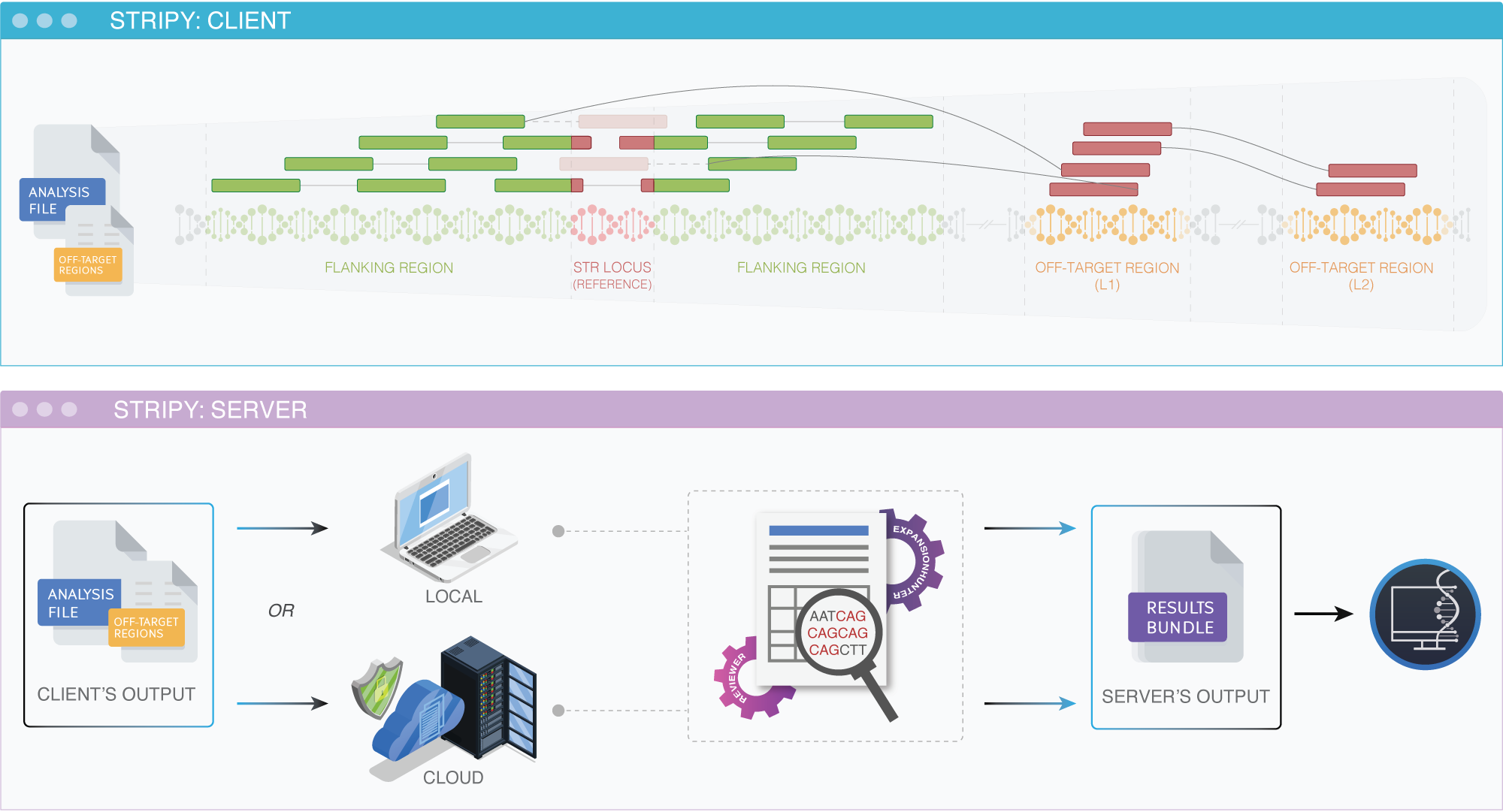
Results are then extracted out from ExpansionHunter and REViewer outputted files, colourised according to the repeat ranges reported in the literature (please see the description of our colour scheme in the documentation page). In the end, final results are shown to the user: interactive and PDF report in the graphical application and HTML and JSON file in the pipeline version. STRipy's graphical application can be configured by clicking on the button on the bottom right corner in the app. STRipy-pipeline requires modifying config.json file in the root folder of the tool.
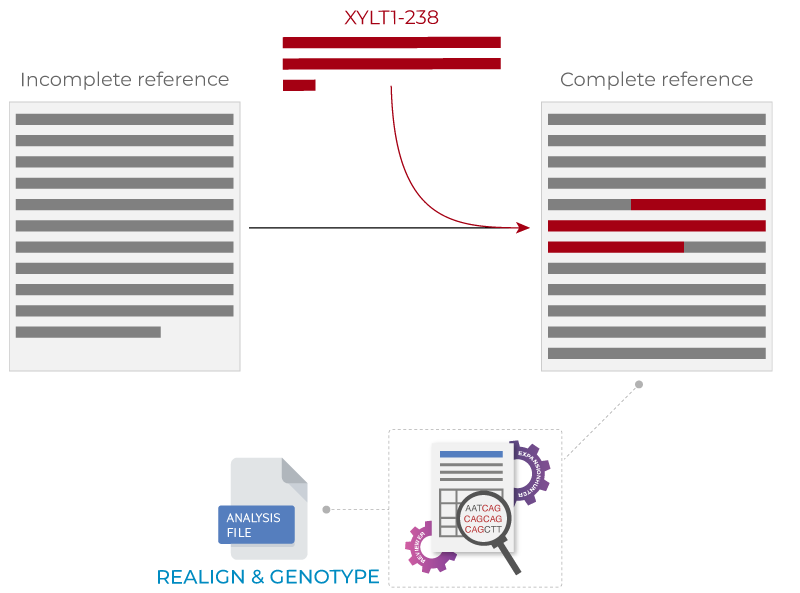
Genotyping reference-missing XYLT1 locus
Repeat expansion in XYLT1 promoter region is known to cause Desbuquois dysplasia type 2 (also known as the Baratela-Scott syndrome). However, the region where the repeat expansion was discovered, is not in the Human reference genome (Faust, et al, 2014) and therefore it has been difficult to genotype the locus in an accurate way. STRipy contains functionality which aims to genotype the locus in an accurate way by realigning reads to a new reference genome which contains a part of the XYLT1 gene and the reference-missing 238 bp sequence.
The inserted sequence is as follows (plus strand, targeted STR region underlined):
CGCCGCCTCGGCTCGCCGCTGCTCCTCCTCCGCCGCCGCCGCCGCCGCTGCCGCCGCCGCCGCCGCCGCCGCCGCCTCCACCGCCGCGGCGCGGAGTTTTCAGACGGGCAGGGACCCGG
ACGTCACCAGGAGGAGGAGAAGGCGGGAGGCGGGAGCGGGGAGGCCGCGGAGGGGGGCGCCGGGCGCGCGCTCGGGGACGGGGCGCGCAGGGAGGGGCGGGCGCCTGGCCCGCGCGGCG
Validation of known pathogenic loci
We validated a locus in each gene by simulating heterozygous as well as homozygous samples where alleles ranged from 60 bp to over 2 kp, using over 60 thousand samples. We simulated 150 bp reads and 450 bp fragments (50 bp SD) and calculated Root Mean Square Error (RMSE) as a measure of accuracy for samples whose long allele was either in one of these three regions: (A) up to read length, (B) from read length to fragment length and (C) over the fragment length. Additionally, we used STRipy on true-positive whole genome samples to confirm its use on real biological samples. STRipy determined an allele to be in the pathogenic range for eight out of nine affected individuals.

STRs database and population-wide data
STRs database has been compiled by using data obtained from the literature. All reference coordinates has been manually curated for each reference genome and presented in 0-based coordinate system. Therefore, the length of a STR locus can differ between reference genomes (for example, hs1 or telomere-to-telomere reference genome has 22 trinucleotide repeats in a row in the DMD locus while hg38 genome has 16 repeats). The end of a STR region was set where either a) repeated sequence ended or b) where there were more than one repeat of another motif (thereby, the STR loci can contain interruptions).
We use repeat ranges derived from the literature for colouring alleles, but it is important to note that STRs are highly complex and these ranges are only indicative, not definite (e.g. pathogenic range might vary between populations, interruptions can affect the pathogenicity, and so on). Population data was obtained by genotyping over two and half thousand whole genome samples in the DRAGEN reanalysis of the 1000 Genomes Project dataset (1kGP-DRAGEN). We used the ExpansionHunter with our own created catalogue to genotype all samples, except for XYLT1 which we used custom made XYLT1 STR caller tool. Replaced and nested types of repeats were genotyped separately by using ExpansionHunter's algorithm only for spanning reads and specifically counting the pathogenic repeat unit. Alleles which were supported by less than 5 spanning reads or less than 50 flanking and in-repeat reads combined were filtered out. This dataset contains phenotypically healthy individuals at the time of genome sequencing, however, some of them could have disease-causing genetic mutations which may manifest at later age.
In the database we have defined repeat types as follows:
- Standard: repeats where one specific motif (for example, CAG or CCG) is in healthy individuals and its expansion becomes pathogenic at larger numbers of repeats.
- Imperfect GCN: repeats which encodes for the amino acid alanine, but the sequence can be composed of different, but synonymous, repeat units – either GCA, GCG, GCC or GCT.
- Replaced/Nested: repeats where the pathogenic motif is not present in the reference genome and mainly not found in healthy individuals. CANVAS is the only example of replaced type repeats, where a stretch made of AAAAG repeats is replaced with a sequence composed of another motif (AAGGG). All the other diseases in this group are caused by nested type of repeats where a stretch made of the pathogenic motif (such as TTTCA) is inserted between or next to the non-pathogenic endogenous repeats (such as TTTTA).
Online tools: ExpansionHunter's catalogue creator and results analyser
We have created two tools for assisting current or previous users of ExpansionHunter and/or DRAGEN with large-scale STRs genotyping. One of the tool we created is ExpansionHunter's catalogue creator which uses our defined loci from the databases and enables to create a custom variant catalogue for ExpansionHunter in a very easy way, just by selecting the loci you want to target. Additionally, off-target regions for all loci can be added to the catalogue. In particular, we simulated long repeated alleles and determined the most likely mis-mapped locations of fully repeated reads in the hg38 reference genome. These coordinates were defined as off-target regions for hg38 and also lifted-over to hg19 and hs1 genome by using UCSC Lift Genome Annotations too by using "minimum ratio of bases that must remap" value 0.95 for hg19 and 0.1 for hs1. This enables genotyping alleles longer than the fragment length. However, it is important to note that one should be cautious when using ExpansionHunter with off-target regions on real-life samples as this could lead to overestimating long alleles when there are fully repeated reads present in the off-target locus which are not originated from the targeted STR locus. Repeats such as CAG which are abundant in the genome can be problematic, while some others like the ones involved in CANVAS are probably less likely. You might want to double-check a sample with STRipy that uses stricter rules for fully repeated reads to minimise overestimation.
The other online tool is ExpansionHunter's results analyser which takes ExpansionHunter's (or DRAGENs STR analysis) JSON results file as an input and then graphically outputs genotypes for each locus which are coloured by whether an allele is in normal, intermediate or pathogenic range. This helps to easily assess your results and it works the best with our catalogue creator due to matching names of loci. NB! Repeat ranges are derived from the literature and therefore they are indicative, not definite (e.g. there could be interruptions present in a locus of your sample which is not evident from the genotype and therefore might not cause the phenotype even when it falls to the pathogenic region). We also mark alleles that are population outliers (P < .0001).
Contact
To contact the author, please use the contact form here. Feel free to ask questions or report any issues with STRipy, our database or online tools. Alternatively you can also send a mail to . Thank you!